Serious vs Non-Serious Adverse Events: When to Report in Clinical Trials
 Mar, 23 2026
Mar, 23 2026
When you're involved in a clinical trial, whether as a participant, researcher, or coordinator, you hear the term adverse event a lot. But not all adverse events are the same. Some are minor, like a headache or mild nausea. Others can be life-changing-or even life-threatening. The difference between a serious and non-serious adverse event isn’t about how bad it feels. It’s about what it does to the person’s health. And knowing which is which determines whether you have to report it right away-or if you can wait until your next routine check-in.
What Makes an Adverse Event "Serious"?
A serious adverse event (SAE) isn’t defined by how intense the symptom is. A severe headache might be painful, but if it doesn’t lead to hospitalization, death, or lasting harm, it’s not serious under regulatory rules. The definition is strict and outcome-based. According to the U.S. Food and Drug Administration (FDA) and the International Council for Harmonisation (ICH), an adverse event is serious if it meets any one of these six criteria:
- It results in death.
- It’s life-threatening (meaning the person was at immediate risk of dying).
- It requires hospitalization or extends an existing hospital stay.
- It causes permanent disability or significant loss of function.
- It leads to a birth defect or congenital anomaly.
- It requires medical or surgical intervention to prevent one of the above outcomes.
That’s it. No gray area. If it doesn’t hit one of these, it’s not serious-even if the patient was in excruciating pain. A study at the University of California, San Francisco found that over 40% of adverse event reports submitted in 2022 were later found to be misclassified. Many of those were cases where researchers confused "severe" with "serious." For example, a patient with cancer experiencing intense fatigue might be described as having a "severe" symptom. But unless that fatigue led to hospitalization or permanent weakness, it doesn’t count as a serious event.
When Do You Have to Report a Serious Adverse Event?
If an event meets the seriousness criteria, the clock starts ticking. Investigators on a clinical trial must report serious adverse events to the sponsor within 24 hours of becoming aware of it. This deadline applies whether or not the event is believed to be caused by the drug or device being tested. The rule is simple: if it’s serious, report it fast.
The sponsor then has its own deadlines. If the event is life-threatening, the sponsor must report it to the FDA within 7 days. For non-life-threatening serious events, the deadline is 15 days. These timelines aren’t suggestions-they’re legal requirements under 21 CFR 312.32. Missing them can lead to regulatory action, delays in trial approval, or even suspension of the study.
But what about the Institutional Review Board (IRB)? The IRB oversees the safety of participants at each research site. Serious adverse events must be reported to the IRB within 7 days. Non-serious events? Often, they’re not reported to the IRB at all unless the study protocol says otherwise. Many sites only review non-serious events during their routine annual or quarterly reviews.
What About Non-Serious Adverse Events?
Non-serious adverse events are the most common. They include things like mild dizziness, temporary rash, or slight upset stomach. These events don’t meet the six seriousness criteria. They might be uncomfortable, but they don’t threaten life or function.
Reporting for these is much less urgent. Instead of a 24-hour clock, they follow the study’s protocol. Most trials collect non-serious events in Case Report Forms (CRFs) and summarize them monthly or quarterly. These reports go to the sponsor and the Data and Safety Monitoring Board (DSMB), which watches for patterns over time. A single mild rash isn’t a red flag. But if 20 participants develop the same rash within a week, that’s a signal worth investigating.
One of the biggest problems in clinical research is that too many non-serious events get reported as serious. A 2020 report from the Clinical Trials Transformation Initiative found that nearly 37% of events labeled as serious by investigators didn’t actually meet the criteria. This floods regulatory systems with noise. The FDA’s Sentinel Initiative has processed over 14 million adverse event reports since 2008. Only about 18% of those were serious. The rest? Mostly minor symptoms that shouldn’t have triggered an urgent report.

Why the Confusion Between Severity and Seriousness?
It’s easy to mix up "severe" and "serious." People hear "severe pain" and assume it’s serious. But in clinical safety, they’re two different things.
Severity describes intensity: mild, moderate, or severe. Seriousness describes outcome: death, hospitalization, disability, etc. A patient could have a severe (intense) migraine that lasts three hours and goes away with ibuprofen. That’s not serious. Another patient could have a mild (slight) rash that turns into toxic epidermal necrolysis-a rare, deadly skin reaction. That’s serious, even though the initial symptom was mild.
Dr. Robert Temple, former FDA Deputy Director, called this confusion "one of the most persistent errors" in clinical safety reporting. It’s not just a paperwork issue-it wastes time, money, and attention. A 2022 survey of 347 research sites found that over 60% had inconsistent seriousness determinations across different studies. In cancer trials, where patients often have baseline symptoms from their disease, the confusion was even worse: 78% of sites had mixed standards.
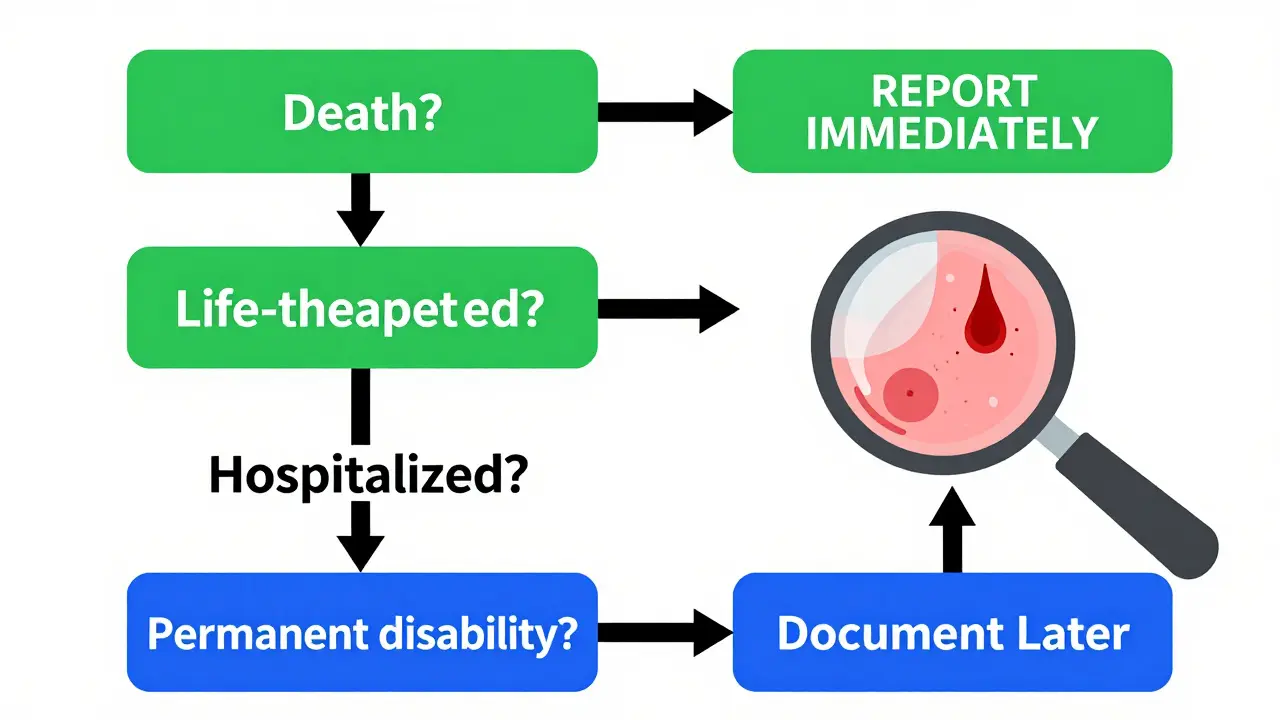
How to Get It Right: A Simple Decision Tree
The National Institute on Aging (NIA) offers a clear, four-question decision tree to determine seriousness. If you’re unsure whether an event is serious, ask:
- Did the event cause death?
- Was the event life-threatening?
- Did it require hospitalization or extend a hospital stay?
- Did it cause persistent or significant disability?
If the answer is "yes" to any of these, it’s serious. Report it immediately. If the answer is "no" to all four, it’s non-serious. Document it in the CRF, but don’t rush the report.
Most sponsors now use the Common Terminology Criteria for Adverse Events (CTCAE) to grade severity (mild, moderate, severe) and apply the ICH/FDA criteria separately for seriousness. This two-step system reduces errors. AI tools are also helping-systems that automatically scan reports for keywords like "hospitalized," "ICU," or "death" now correctly classify seriousness in nearly 90% of cases. But they still need human review. Machines can’t always tell if a "hospital visit" was for a routine check-up or an emergency.
What’s Changing in 2026?
The system is evolving. In 2023, the FDA proposed new guidance to standardize seriousness criteria across all disease areas. Right now, a heart attack in a heart study might be treated differently than one in a diabetes trial. The new rules aim to fix that.
The European Union’s Clinical Trials Regulation, fully in force since 2022, has already harmonized definitions across all 27 member states. That’s cut down reporting errors by over a third. The FDA is now testing AI tools that use natural language processing to auto-triage reports. Early results show a 47% reduction in processing time.
Training is also getting stricter. Nearly all major research institutions now require annual safety training for staff. The goal? Make sure everyone understands the difference between "severe" and "serious." Because when you report the wrong thing, you don’t just waste time-you risk missing a real danger.
Bottom Line
When you’re dealing with adverse events in clinical research, don’t guess. Don’t rely on how bad it sounds. Use the six criteria. If it’s not one of them, it’s not serious. Report non-serious events as scheduled. Report serious events within 24 hours. That’s the rule. And in safety monitoring, the rule isn’t just paperwork-it’s what keeps patients alive.
Zola Parker
March 24, 2026 AT 15:33Elaine Parra
March 25, 2026 AT 08:45Natasha Rodríguez Lara
March 26, 2026 AT 06:31peter vencken
March 27, 2026 AT 22:30James Moreau
March 29, 2026 AT 12:36J. Murphy
March 30, 2026 AT 12:40Jesse Hall
March 31, 2026 AT 22:34Donna Fogelsong
April 1, 2026 AT 21:51Sean Bechtelheimer
April 2, 2026 AT 12:34